Smart-seq3Xpress: a scalable, high-sensitivity and low-cost Single cell Full-length RNA-seq methodology.
Mar 5, 2024
Share article


Dr. Weilin Liu, Senior Field Application Scientist

Single-cell sequencing, which provides a high-resolution of cellular differences by sequencing nucleic acid information namely DNA and RNA from individual cells, has become one of the key applications in the next-generation sequencing (NGS) to better understand the individual heterogeneity in physiological processes and the personal differences in responding of medical treatments, shedding light on discovering novel biological mechanisms and personalized therapies.
Currently, most single-cell RNA-sequencing (scRNA-seq) methods map a short part of the RNA molecules, from either the 5′ or 3′ end, together with a unique molecular identifier (UMI)(Mereu et al., Nat. Biotechnol, 2020). These RNA end-counting strategies have been efficient in estimating gene expression across large numbers of cells, while controlling for PCR amplification biases attributed to low quantity input of RNAs and compensating on the throughput capacity of sequencing, but the limited coverage of those methods heavily obscure the detection of those transcribed genetic variations and transcript isoform expression caused by, for instance, RNA alternative-splicing, which is a common mechanism of RNA processing and abnormalities in such a process have been shown to associate with the development of various diseases, such as cancers and neurodegenerative diseases (Zhang et al., Nature, 2021; Garcia-Blanco et al., Nat. Biotechnol, 2004; Mills et al., Neurobiol Aging, 2012). Moreover, though those end-counting scRNA-seq enable higher throughput sequencing capacity in terms of cell number, their relatively low sensitivity and high cost are the major bottlenecks to be challenged. In the meanwhile, long-read sequencing technologies enable direct quantification of allele- and isoform-level expression, however their current read depths, biased-error rate and sample preparation procedures obstruct their broad applications across cells, tissues and organisms (Byrne et al., Nat. Commum, 2017; Gupta et al., Nat. Biotechnol, 2018)
To overcome those technical demerits, Hagemann-Jensen and Ziegenhain et al. at Karolinska Institute (Nat. Biotechnol, 2022; Nat. Biotechnol, 2020. https://www.nature.com/articles/s41587-022-01311-4) have developed and further optimized a scalable and sensitive short-read based sequencing method, Smart-seq3xpress to enable full-length RNA coverage and more importantly to provide an unprecedent high-resolution of individual RNAs to isoforms and their alleic origin in single cells with lower operating cost and shortened library preparation duration to a single workday. The well-established Smart-seq3xpress protocol implements UMI in the 5’-end of full-length RNA transcripts to establish a unique identity of each RNA molecule, thus inclusion of UMI empowers the detection of transcript copy number variation (CNV) and to overcome the biases caused by PCR amplification processes, further bringing up sequencing sensitivity and specificity. Moreover, Smart-seq3xpress is a plate-based method, which could be easily scaled up from 96- to 384-well plates possibly facilitated by automation processes. More practically, sorting of the cells of interest into the well-plates can be separated in spatial and temporal manner as the filled well-plates can be frozen anytime until further processing, showing a major advantage when compared to the method that viable cell suspension needs to be processed immediately. Moreover, this study also put substantial effort in miniaturizing the key factors in scRNA-seq, including lowering the reaction volume, decreasing the input of cDNA, reducing PCR cycles, and adjusting Tn5 and cDNA ratio for more cost-effective tagmentation without compromising the detection performance. Additionally, adjusting of the salt components and concentration required for the reactions, implementation of new enzymes and uniquely setting an extensive RNA count QC by utilizing of self-developed new UMIcountR R package to further improve the overall detection power of Smart-seq3xpress. Furthermore, this method provides a more environmental-friendly and cost-effective solution because the material and resources are ten-fold less when compared with other RNA-seq methods, and the usage plastics consumables is significantly reduced by new 3D-printed tools and by contact-less reagent dispensing as well as pre-dispensed desiccated index primers`.
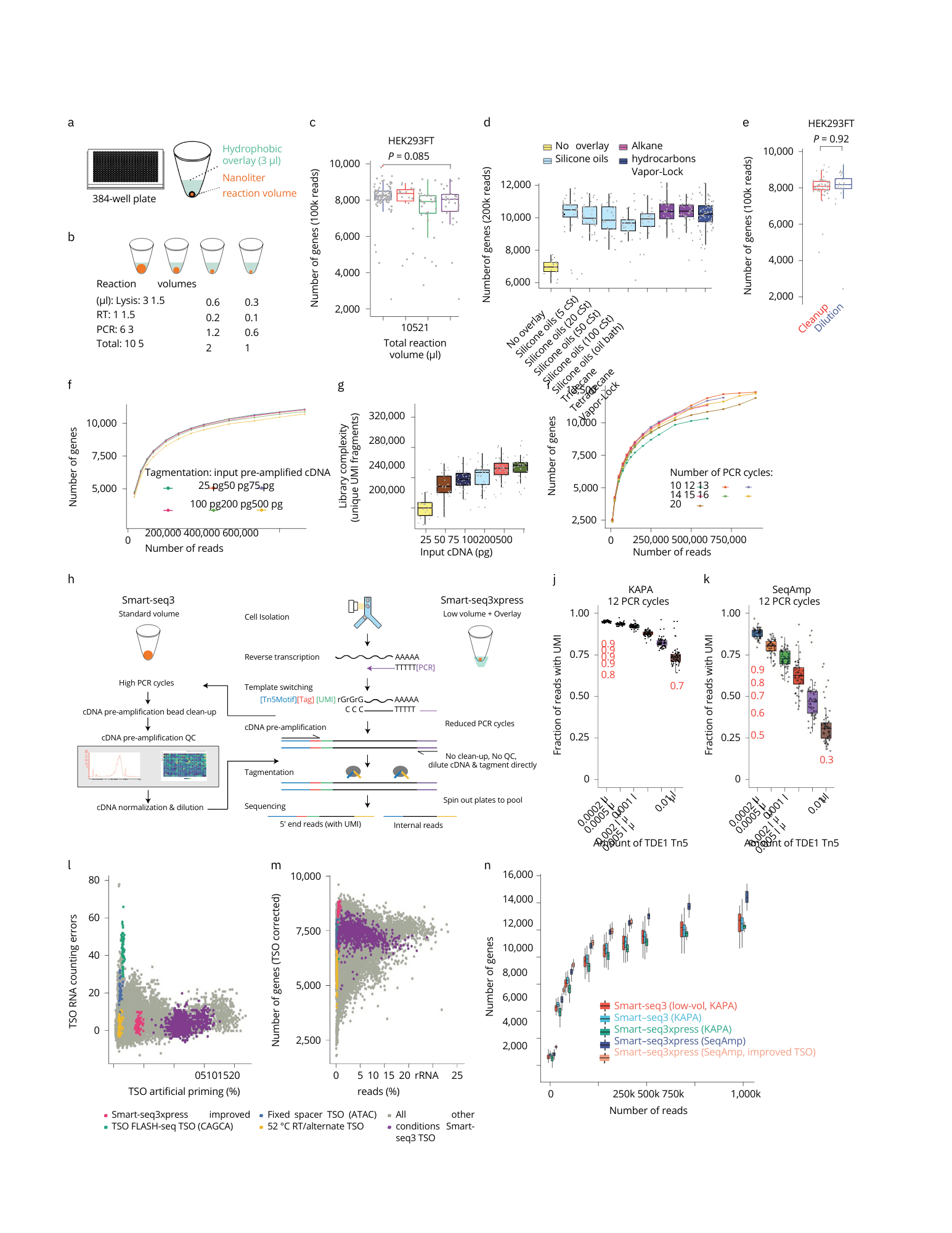
fig. 1 | Scalable full-transcript coverage scRNA-seq with Smart-seq3xpress. a, Schematic of nanoliter cDNA synthesis reactions performed in wells of 384-well PCR plates with 3 µl of hydrophobic overlay. b, Illustration of reduced-volume experiments with the lysis, RT and PCR volumes used. c, The number of genes detected per HEK293TF cell at each reaction volume, when sampling 100,000 sequencing reads (n= 100, 19, 32 and 28 cells, respectively). P value represents a two-sided t-test between the 10-µl and 1-µl conditions. d, Influence of hydrophobic overlays on miniaturized cDNA synthesis (1 µl total volume). For each compound, boxes depict the number of genes detected per HEK293FT cell (n= 17, 34, 39, 28, 25, 24, 28, 38 and 70, respectively), subsampled at 200,000 sequencing reads per cell. e, Replacement of the bead-based cDNA cleanup by dilution in single HEK293FT (n= 58 and 52, respectively) cells. Box plots show the number of genes detected per cell and condition (at 100,000 reads) with P value for a two-sided t-test across conditions. f, Tagmentation complexity using 0.1 µl of ATM Tn5 enzyme per HEK293FT cell in relation to input cDNA. The median number of detected genes as a function of raw sequencing reads (n= 51, 53, 54, 53, 53 and 52 cells for 25, 50, 75, 100, 200 and 500 pg, respectively). g, Tagmentation complexity for varying amounts of cDNA input. Complexity was summarized as unique aligned and gene-assigned UMI-containing read pairs per 400,000 raw reads and HEK293FT cell (n= 49, 51, 51, 50, 51 and 44). h, Schematic outline of the Smart-seq3 and Smartseq3xpress workflows. i, The number of genes detected with Smart-seq3xpress after variable amounts of pre-amplification PCR cycles. Median number of genes is reported as a function of raw sequencing reads in HEK293FT cells (n= 93, 98, 108, 113, 102, 114 and 118 cells for 10, 12, 13, 14, 15, 16 or 20 cycles, respectively). j, Fraction of UMI-containing reads to internal reads for HEK293FT cells prepared with Smartseq3xpress (KAPA HiFi; 12 PCR cycles), at a variable range of TDE1 Tn5 amounts (n= 64 cells each). k, Fraction of UMI-containing reads to internal reads for HEK293FT cells prepared with Smartseq3xpress (SeqAmp; 12 PCR cycles), at a variable range of TDE1 Tn5 amounts (n= 60 cells each). l,m, Optimization of RT and PCR conditions across 376 experimental conditions on HEK293FT cells. Colors indicate particular experimental conditions: Smart-seq3xpress with Smart-seq3 TSO (purple; n= 912), 52 °C RT/alternate TSO implementation (yellow; n= 74), fixed spacer TSO variant (blue; n= 45), FLASH-seq TSO variant (green; n= 55), Smart-seq3xpress with improved TSO (pink; n= 63) and all other conditions (gray; n= 21,707). Scatter plots denote the level of artifactual TSO-UMI reads and RNA counting errors (l) as well as a percentage of ribosomal RNA (rRNA) mapped reads and number of detected genes in 100,000 reads after removal of strand invasion reads (m). n, Benchmarking of Smart-seq3 variants. Box plots show the number of genes detected per HEK293FT cell in full-volume Smart-seq3 (ref. 2), low-volume Smart-seq3 and Smart-seq3xpress implementations, at the indicated read depths (n= 109–110, 18–27, 9–170, 20–55 and 9–63 cells, depending on the cells available at the given sequencing depths). The box plots (in c, d, e, j, k and n) show the median and first and third quartiles as a box, and the whiskers indicate the most extreme data points within 1.5 lengths of the box. cSt, centistoke.
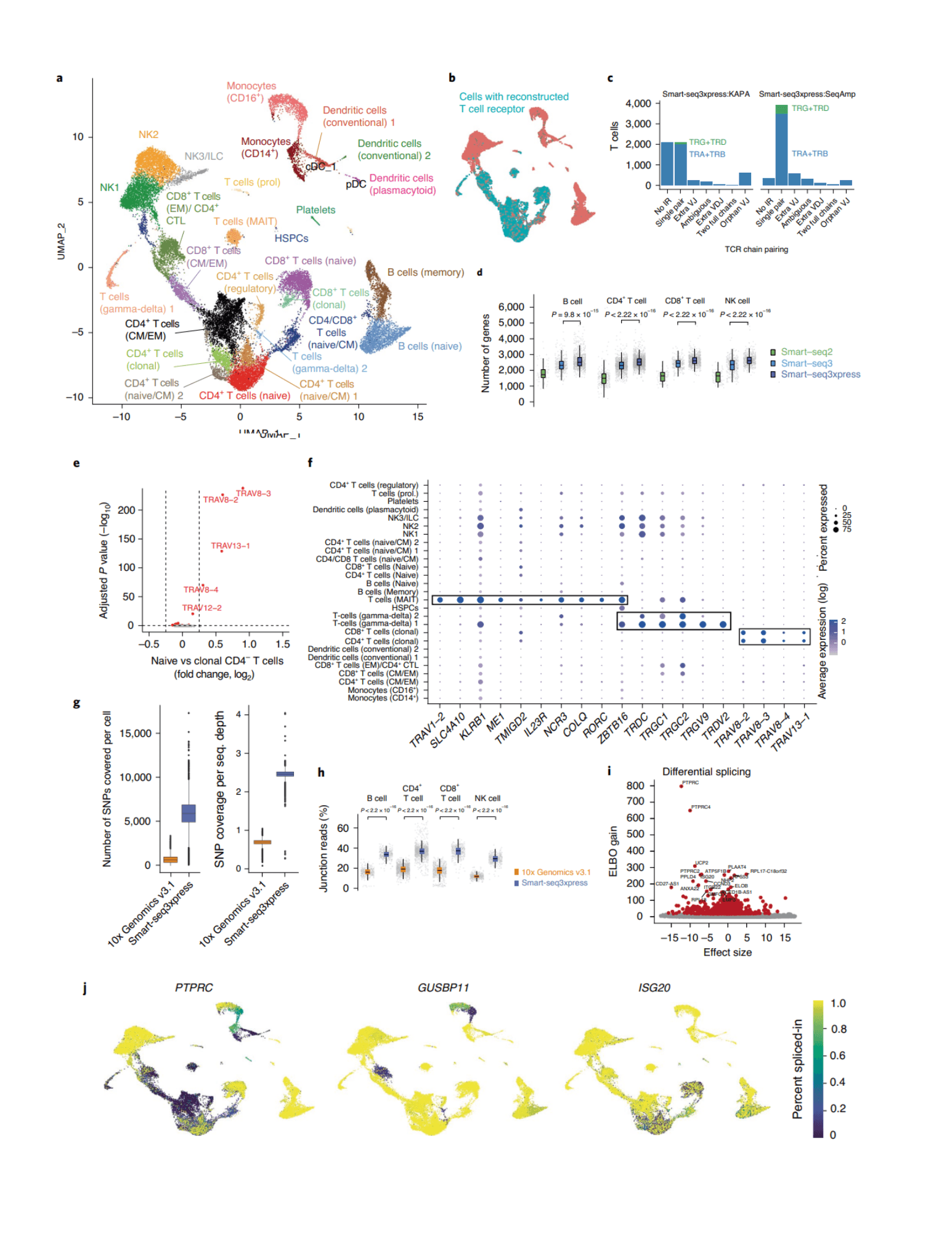
To demonstrate Smart-seq3xpress, Hagemann-Jensen and Ziegenhain et al. examined 26,260 human peripheral blood mononuclear cells (hPBMCs) with an average depth of 258,000 read pairs per cell. The results provided a full-length transcript profiling and showed the reconstruction of T-cell receptor sequences, identifying several cell types and states, including unconventional T-cell populations such as MAIT, and gamma-delta T-cells, with higher gene detection across cell types. Notably, these results are achieved by using almost 10 times less hPBMCs when compared to the droplet-based sc-RNAseq method. In the following comparison study with a droplet-based method, Smart-seq3xpress gave more superior performances in multiple aspects in terms of single-nucleotide polymorphism (SNP) detection (9-fold higher), more read support over exon–exon/exon–intron splice junctions by its full-length RNA coverage and T-cell clustering consistency. The study of hPBMC Smart-seq3xpress data on alternative splicing further revealed significant variation in inclusion levels and distinct splicing patterns among cell types, of which are independent from their gene expression level. A later benchmarking study (Probst et al., BMC Genomics, 2022) on full-length scRNAseq indicated that Smart-seq3 protocol presented the highest gene detection per single cell at the lower price when compared with other kits.
About MGI
MGI Tech Co., Ltd. (MGI) is at the forefront of global innovation, actively contributing to life science through intelligent innovation. With a presence in over 100 countries and a customer base of 2600+, MGI's cutting-edge technology has been instrumental in the development of 736+ user patents, facilitating the creation of over 150 petabytes of data. The company's extensive portfolio includes sequencing instruments, automation instruments, reagents, and related products that cater to various sectors such as life science research, agriculture, precision medicine, and healthcare.
MGI is dedicated to advancing life science tools for the healthcare of the future. As of December 2021, the company's global presence has expanded to more than 100 countries and regions, serving over 1,300 international clients. With a workforce of over 2,900 professionals worldwide, including 5 centers for research and development and production facilities in Europe, MGI is committed to fostering innovation. Approximately 35% of MGI's employees are engaged in R&D, underscoring the company's focus on pioneering advancements in the field.
The impact of MGI's work is further evidenced by the publication of over 6,800 papers in prestigious scientific journals, showcasing the significant contribution of MGI's technology to the scientific community and beyond.
For more information about MGI and its contributions to life science and healthcare, please visit the MGI website or connect with us on Twitter, LinkedIn, or YouTube.
Advancements in Single-Cell Sequencing
Smart-seq3xpress
Pioneering Full-Length scRNAseq:
hPBMC Studies
Share this article :
Share
More Science

Jan 10, 2024
Single-cell Spatial transcriptomics (sc-StereoSeq), opening a new era in plants and crops biology.
Explore how sc-StereoSeq is transforming our understanding of plant biology, from crop production to plant stress responses, marking a new era in spatial transcriptomics.

Oct 10, 2023
Can Next-generation Sequencing accelerate mRNA vaccine development?
Explore the transformative impact of Next-Generation Sequencing in mRNA vaccine development. Dive into Nobel-winning discoveries, challenges, and novel technologies like the VAX-seq workflow, enhancing vaccine production, safety, and quality analysis amidst global health crises.

Sep 1, 2023
A Complete Human Y to ask more "Why" in biology
Unlock the mysteries of the human Y chromosome with MGI's cutting-edge findings. Learn how the T2T consortium's study corrects GRCh38 errors, reveals 30Mb+ new sequence data, and advances our understanding of gene structures and their implications in the Omic-verse.